지난해 시판 승인을 받은 복제약(제네릭) 중 다른 업체에서 위탁 생산하는 제품 비중이 80%를 넘어서는 것으로 나타났다. 위탁 의약품 허가 요건 완화로 직접 제네릭 개발을 위한 임상시험을 진행하는 것보다 다른 업체의 생산시설을 활용해 제네릭 시장에 진출하는 사례가 급증하고 있다.
21일 식품의약품안전처 식품의약품안전평가원은 지난해 제네릭 의약품 허가를 위한 생물학적동등성시험(생동성시험) 계획 승인 건수가 123건으로 전년 대비 약 39% 감소했다고 밝혔다.
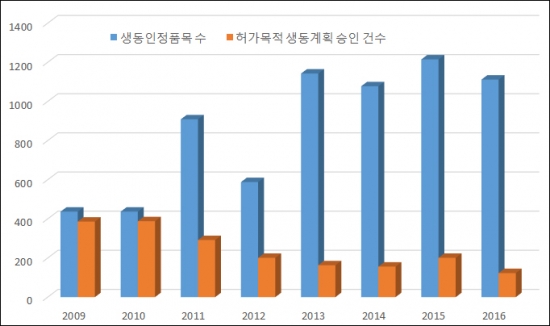
허가 목적 생동성시험 승인 건수는 2009년 386건에서 지속적으로 감소세를 나타내다 7년 동안 3분의 1 수준으로 줄었다. 제약사들이 제네릭 허가를 위한 생동성시험 시도가 크게 감소했다는 의미다.
그렇지만 제네릭 허가 건수가 줄어든 것은 아니다. 생동성시험을 마치고 시판승인을 받은 ‘생동성 인정 품목’ 건수는 지난해 1112건으로 2009년 437건보다 2.5배 늘었다. 매년 허가 목적 생동성시험 시도가 큰 폭으로 감소하는 것과 달리 허가받은 제네릭은 급증세를 나타낸 셈이다.
생동성시험 계획을 승인받고 제네릭 제품이 최종 시판승인을 받을 때까지 일정 기간의 시차가 발생하는 것을 감안하더라도, 허가 목적 임상시험과 허가 건수가 반비례하는 기현상이 연출된 것이다.
제네릭 허가 진입 장벽이 낮아진데 따른 변화로 분석된다.
우선 지난 2011년 11월 공동 생동 규제가 폐지된 이후 다른 업체에서 제네릭을 생산해 허가받는 ‘위탁 제네릭’이 크게 늘었다.
당초 식약처는 같은 공장에서 생산한 제네릭이더라도 생동성시험은 2개사까지만 함께 진행토록 제한하는 공동(위탁)생동 제한 규정을 시행했다.
같은 공장에서 똑같은 공정을 통해 같은 성분의 6개 제네릭을 생산하더라도 생동성시험은 2개씩 3그룹으로 나눠서 진행해야 한다는 의미다. A제약사가 B사가 생산중인 제품을 제품명만 다르게 위탁받아 생산하고 허가를 받을 때에도 A사가 별도로 생동시험을 진행해야만 허가를 내줬다. ‘같은 공장에서 생산하는 똑같은 의약품인데도 임상시험을 따로 진행하토록 하는 것은 불합리한 규제’라는 제약업계의 지적에 2011년 이 규제는 사라졌다.
여기에 지난 2014년 의약품을 생산하는 모든 공장은 3년마다 식약처가 정한 시설기준을 통과해야 의약품 생산을 허용하는 내용의 ‘GMP 적합판정서 도입’이라는 새로운 허가 제도를 시행하면서 허가용 의약품을 의무적으로 생산해야 하는 규정이 큰 폭으로 완화됐다.
기존에는 다른 업체가 대신 생산해주는 위탁 의약품이 허가를 받으려면 3개 제조단위(3배치)를 미리 생산해야 했지만 적합판정을 통과한 제조시설에서 생산 중인 제네릭을 제품명과 포장만 바꿔 허가받을 때 절차가 간소화됐다.
이에 따라 제약사 입장에서는 허가 신청 수수료 100만원 안팎만 부담하면 별도의 생동성시험과 허가용 의약품 생산 절차를 거치지 않고도 위탁 제네릭 허가가 가능해졌다.
만약 매년 생동성시험 계획을 승인받은 제품이 이듬해 시판허가를 받는다고 가정하면(통상 생동성시험 계획 승인부터 시판허가까지 9~12개월 가량 소요), 2009년 386개 품목이 허가목적으로 생동시험 계획을 승인받은 이후 2010년에 437건의 제네릭 허가가 이뤄졌다. 허가받은 제네릭의 88%는 직접 생동성시험을 진행했다는 의미다.
이에 반해 2015년 201건의 생동성시험계획이 승인받았는데, 지난해 5.5배 많은 1112건의 제네릭이 시판 허가를 받았다. 지난해 허가받은 제네릭 5개 중 4개는 직접 생동성시험을 진행하지 않은 위탁제네릭이라는 추정이 나온다.
식약처 관계자는 “생동성시험을 여러 제약사가 공동으로 진행할 수 있게 되고 수익성이 높은 제품의 제약사간 공동 개발이 증가하면서 생동성시험 계획 승인 건수가 지속적으로 감소했다”라고 설명했다.
한편 지난해 생동성시험 계획 승인을 받은 123건 중 베링거인겔하임의 ‘트라젠타(성분명 리나글립틴)’의 제네릭이 18건으로 가장 많았다. ‘베포타스틴’ 성분의 알레르기 비염치료제(15건), ‘티카그렐러’ 성분의 혈전성심혈관질환 치료제(10건) 제네릭 개발을 위한 생동성시험이 뒤를 이었다.
![부동산 PF 체질 개선 나선다…PF 자기자본비율 상향·사업성 평가 강화 [종합]](https://img.etoday.co.kr/crop/140/88/2101720.jpg)
![‘2025 수능 수험표’ 들고 어디 갈까?…수험생 할인 총정리 [그래픽 스토리]](https://img.etoday.co.kr/crop/140/88/2102319.jpg)
![전기차 수준 더 높아졌다…상품성으로 캐즘 정면돌파 [2024 스마트EV]](https://img.etoday.co.kr/crop/140/88/2102282.jpg)
![낮은 금리로 보증금과 월세 대출, '청년전용 보증부월세대출' [십분청년백서]](https://img.etoday.co.kr/crop/140/88/2102283.jpg)
![[종합] ‘공직선거법 위반’ 김혜경 벌금 150만원…法 “공정성·투명성 해할 위험”](https://img.etoday.co.kr/crop/140/88/2102280.jpg)
![이혼에 안타까운 사망까지...올해도 연예계 뒤흔든 '11월 괴담' [이슈크래커]](https://img.etoday.co.kr/crop/140/88/2101314.jpg)
!["늦었다고 생각할 때가 제일 빠를 때죠" 83세 임태수 할머니의 수능 도전 [포토로그]](https://img.etoday.co.kr/crop/140/88/2101767.jpg)












![‘2025 수능 수험표’ 들고 어디 갈까?…수험생 할인 총정리 [그래픽 스토리]](https://img.etoday.co.kr/crop/300/170/2102319.jpg)
![본회의, 이재명 대표 '표정'…1심 재판 선고 D-1 [포토]](https://img.etoday.co.kr/crop/300/190/2102368.jpg)